Definition:
Polycythemia is the increase of the RBC count, hemoglobin, and total RBC volume, accompanied by an increase in total blood volume. This must be distinguished from relative erythrocytosis secondary to fluid loss or decreased intake; this distinction can be made easily on a clinical basis. Polycythemia accompanies increased total blood volume, whereas relative erythrocytosis does not. Two basic categories of polycythemia are recognized:
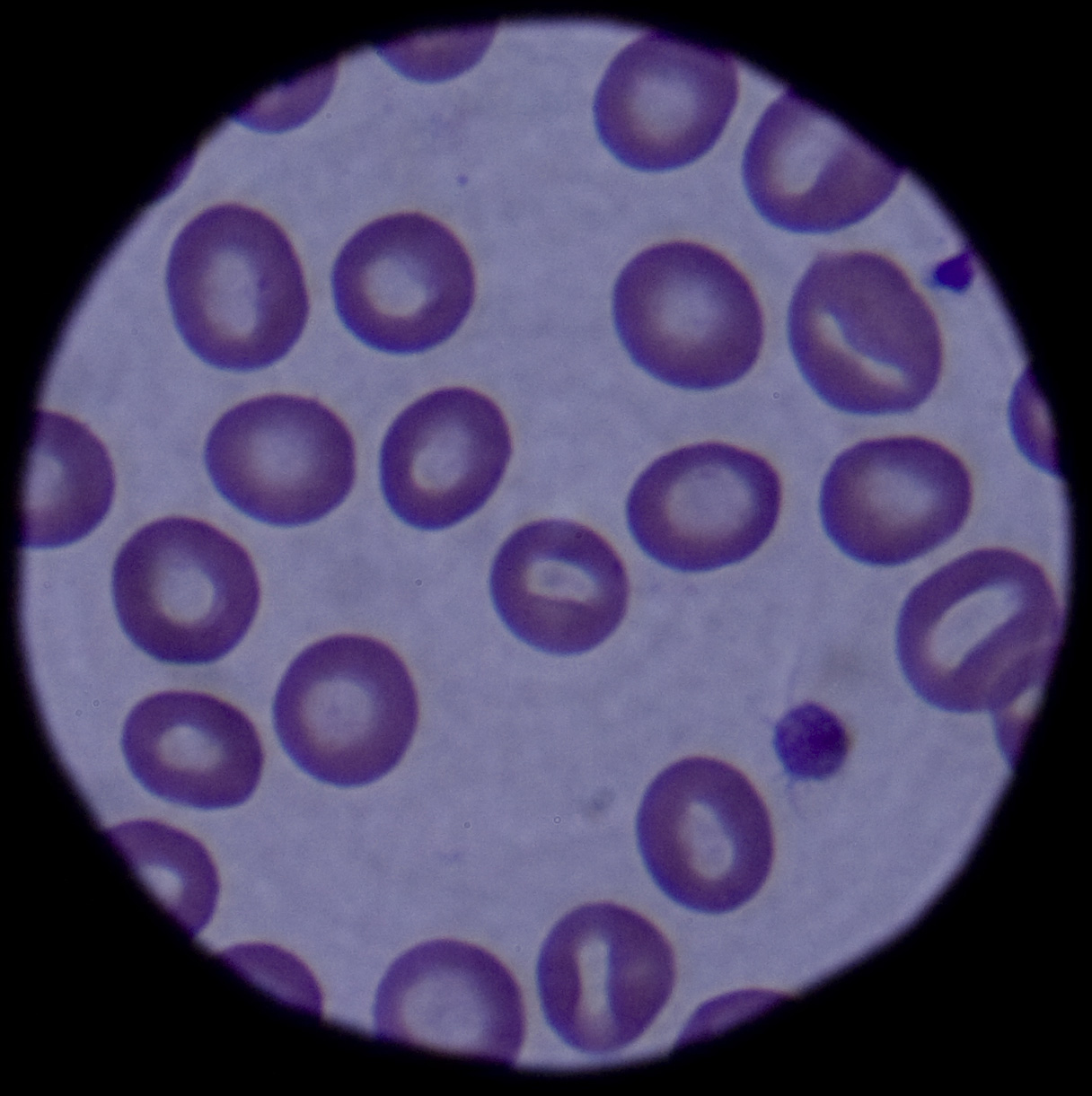
...click to see…the picture..
* Primary polycythemias are due to factors intrinsic to red cell precursors and include the diagnoses of primary familial and congenital polycythemia (PFCP) and polycythemia vera (PV).
* Secondary polycythemias are caused by factors extrinsic to red cell precursors.
In normal hematopoiesis, myeloid stem cells give rise to erythrocytes, platelets, granulocytes, eosinophils, basophils, and monocytes. The production of each lineage is a function of cell proliferation, differentiation, and apoptosis. These various stages of differentiation rely on multiple interrelated processes. Protein growth factors, known as cytokines, stimulate proliferation of the multilineage cells (eg, interleukin [IL]-3, granulocyte-macrophage colony-stimulating activity [GM-CSF]). Other factors primarily stimulate the growth of committed progenitors (eg, GM-CSF, macrophage colony-stimulating factor [M-CSF], erythropoietin [Epo]).
Erythropoiesis is a carefully ordered sequence of events. Initially occurring in fetal hepatocytes, the process is taken over by the bone marrow in the child and adult. Although multiple cytokines and growth factors are dedicated to the proliferation of the RBC, the primary regulator is Epo. Red cell development is initially regulated by stem cell factor (SCF), which commits hematopoietic stem cells to develop into erythroid progenitors. Subsequently, Epo continues to stimulate the development and terminal differentiation of these progenitors. In the fetus, Epo is produced by monocytes and macrophages found in the liver. After birth, Epo is produced in the kidneys; however, Epo messenger RNA (mRNA) and Epo protein are also found in the brain and in RBCs, suggesting that some paracrine and autocrine function is present as well. CLICK & SEE THE PICTURES
Erythropoiesis escalates as increased expression of the EPO gene produces higher levels of circulating Epo. EPO gene expression is known to be affected by multiple factors, including hypoxemia, transition metals (Co2+, Ni2+, Mn2+), and iron chelators. However, the major influence is hypoxia, including factors of decreased oxygen tension, RBC loss, and increased oxygen affinity of hemoglobin. In fact, Epo production has been observed to increase as much as 1000-fold in severe hypoxia.
Absolute polycythemia
The overproduction of red blood cells may be due to a primary process in the bone marrow (a so-called myeloproliferative syndrome), or it may be a reaction to chronically low oxygen levels or, rarely, a malignancy.
Primary polycythemia (Polycythemia vera)
Primary polycythemia, often called polycythemia vera (PCV), polycythemia rubra vera (PRV), or erythremia, occurs when excess red blood cells are produced as a result of an abnormality of the bone marrow. Often, excess white blood cells and platelets are also produced. Polycythemia vera is classified as a myeloproliferative disease. Symptoms include headaches, vertigo, and an abnormally enlarged spleen and/or liver. In some cases, affected individuals may have associated conditions including high blood pressure or the formation of blood clots. Transformation to acute leukemia is rare. Phlebotomy is the mainstay of treatment. A hallmark of polycythemia is an elevated hematocrit, with Hct > 55% seen in 83% of cases. Mutations in JAK2 are found in 95% of cases, though also present in other myeloproliferative disorders.
Secondary polycythemia
Secondary polycythemia is caused by either natural or artificial increases in the production of erythropoietin, hence an increased production of erythrocytes. In secondary polycythemia, there may be 6 to 8 million and occasionally 9 million erythrocytes per cubic millimeter (microliter) of blood. Secondary polycythemia resolves when the underlying cause is treated.
Secondary polycythemia in which the production of erythropoietin increases appropriately is called physiologic polycythemia. This physiologic (meaning normal) polycythemia is a normal adaptation to living at high altitudes (see altitude sickness). Many athletes train at high altitude to take advantage of this effect — a legal form of blood doping. Similarly, athletes with primary polycythemia may have a competitive advantage due to greater stamina.
Other causes of secondary polycythemia include smoking, renal or liver tumors, hemangioblastomas in the central nervous system, heart or lung diseases that result in hypoxia, and endocrine abnormalities including pheochromocytoma and adrenal adenoma with Cushing’s syndrome. People whose testosterone levels are high because of the use anabolic steroids, including athletes who abuse steroids and people whose doctors put them on doses that are too high, as well as people who take erythropoietin may develop secondary polycythemia.
Secondary polycythemia can be induced directly by phlebotomy to withdraw some blood, concentrate the erythrocytes, and return them to the body.
Chuvash polycythemia
Chuvash polycythemia refers to a familial form of erythrocytosis different from classical polycythemia vera. This involved patients from Chuvashia and is associated with a C598T mutation in the von Hippel-Lindau gene (VHL).[6] A cluster of patients with Chuvash polycythemia have been found in other populations, such as on the Italian island of Ischia, located in the Bay of Naples.
Relative polycythemia
Relative polycythemia is an apparent rise of the erythrocyte level in the blood; however, the underlying cause is reduced blood plasma. Relative polycythemia is often caused by loss of body fluids, such as through burns, dehydration and stress.
Polycythemia vera
Earlier diagnostic criteria for polycythemia vera included the following (based on the Polycythemia Vera Study Group Diagnostic Criteria):1
* Red cell mass greater than 36 mL/kg for men and greater than 32 mL/kg for women
* Arterial oxygen saturation greater than 92%
* Splenomegaly or 2 of the following:
o Thrombocytosis greater than 400 X 109/L
o Leukocytosis greater than 12 X 109/L
o Leukocyte alkaline phosphatase activity greater than 100 U/L in adults (reference range, 30-120 U/L) without fever or infection
o Serum vitamin B-12 greater than 900 pg/mL (reference range, 130-785 pg/mL)
o Unsaturated vitamin B-12 binding capacity greater than 2200 pg/mL
The reference range for the clinician’s laboratory should be cross-correlated. The diagnostic criteria have undergone scrutiny and several revisions in recent years. In 2001, the World Health Organization (WHO) proposed a classification system for chronic myeloid neoplasms.2 The diagnosis of polycythemia vera fell under the broader category of chronic myeloproliferative diseases. This set of criteria quickly lost favor because of lack of validation3 and the discovery of JAK2 mutations in adult patients.4,5,6,7,8,9
Currently, the diagnosis of polycythemia vera is based on the 2008 WHO criteria, which has integrated molecular diagnostics into the evaluation and screening for polycythemia vera.10 A diagnosis of polycythemia vera is made when both major and one minor criterion are present or when the first major criterion is present with any two minor criteria.
The current criteria include the following:
* Major criteria
1. Hemoglobin level of more than 18.5 g/dL in men (>16.5 g/dL in women) or other evidence of increased red cell volume
or
Hemoglobin or hematocrit level higher than 99th percentile of method-specific reference range for age, sex, altitude, of residence
or
Hemoglobin level of more than 17 g/dL in men (>15 g/dL in women) if associated with a documented and sustained increase of at least 2 g/dL from an individual’s baseline value that can not be attributed to correction of iron deficiency
or
Elevated red cell mass greater than 25% above mean normal predicted value
2. Presence of JAK2V617F or similar mutation (eg, JAK2 exon 12 mutation)
* Minor criteria
1. Bone marrow trilineage myeloproliferation
2. Subnormal serum erythropoietin levels
3. Endogenous erythroid colony growth
Pathophysiology
Primary polycythemia
The disease is considered to be a form of the myeloproliferative syndromes that include polycythemia vera, essential thrombocythemia, agnogenic myeloid metaplasia, and myelofibrosis. The clonality of polycythemia vera is well established and was first demonstrated by Adamson et al in 1976.11 Subsequent studies suggest hypersensitivity of the myeloid progenitor cells to growth factors, including Epo, IL-3, SCF, GM-CSF, and insulinlike growth factor (IGF)–1, whereas other studies show defects in programmed cell death.
Until recently, the pathophysiology of polycythemia vera was unclear. In 2005, significant progress in the understanding of polycythemia vera was made with the discovery of an activating mutation in the tyrosine kinase JAK2 (JAK2V617F ), which now appears to cause most primary cases in adults.4,5,6,7,8 Several other mutations of JAK2 have since been described (eg, exon 12, JAK2H538-K539delinsI ).9,12 The JAK2 mutations are thought to possibly cause hypersensitivity to Epo via the Epo receptor, although the effects of this mutation remain to be fully characterized.
Familial clustering suggests a genetic predisposition. Whether these mutations are responsible for the development of polycythemia vera in pediatric patients is unclear. Some groups have reported lower rates of JAK2 mutations in children compared with adults,13,14,15 whereas other groups have seen similar rates with complete or near complete presence of JAK2V617F and other JAK2 mutations.12
PFCP is caused by a hypersensitivity of erythroid precursors to Epo. Several mutations (approximately 14) have been identified in the Epo receptor (EPOR) gene; however, EPOR mutations have not been identified in all PFCP kindreds. Most identified EPOR mutations (11) cause truncation of the c-terminal cytoplasmic receptor domain of the receptor. These truncated receptors have heightened sensitivity to circulating Epo due to a lack of negative feedback regulation.16
Secondary polycythemia
Secondary polycythemia may result from functional hypoxia induced by lung disease, heart disease, increased altitude (hemoglobin increase of 4% for each 1000-m increase in altitude), congenital methemoglobinemia, and other high–oxygen affinity hemoglobinopathies stimulating increased Epo production. Secondary polycythemia may also result from increased Epo production secondary to benign and malignant Epo-secreting lesions. Secondary polycythemia may also be a benign familial polycythemia.
Chuvash polycythemia, a congenital polycythemia first recognized in an endemic Russian population, has mutations in the von Hippel-Lindau (VHL) gene, which is associated with a perturbed oxygen dependent regulation of Epo synthesis.
Secondary polycythemia of the newborn is fairly common and is a result of either chronic or acute fetal hypoxia or delayed cord clamping and stripping of the umbilical cord.17
Frequency
United States
Primary polycythemia is rare; the overall prevalence of polycythemia vera is 2 cases per 100,000 people. The median age is 60 years. Only 0.1% of cases of polycythemia vera are observed in individuals younger than 20 years. Fewer than 50 cases of pediatric polycythemia vera have been reported in the literature. Secondary polycythemia is seen in 1-5% of all newborns in the United States.
International
Polycythemia vera has a similar incidence in Western Europe as in the United States, and occurrence rates are very low in Africa and Asia (as low as 2 cases per million per year in Japan).
Mortality/Morbidity
Death rates for children are unavailable. The complications found in polycythemia vera are related to 2 primary factors. The first includes complications related to hyperviscosity. The second involves bone marrow–related complications. Untreated, the median survival time for these patients is 18 months. However, if patients are treated, survival is greatly extended, as many as 10-15 years with phlebotomy alone. The causes of death in adults are as follows:
* Thrombosis/thromboembolism (30-40%)
* Acute myelogenous leukemia (19%)
* Other malignancies (15%)
* Hemorrhage (2-10%)
* Myelofibrosis/myeloid metaplasia (4%)
* Other (25%)
In the neonatal period, polycythemia-induced hyperviscosity can lead to altered blood flow and subsequently affect organ function. Infants with polycythemia are at increased risk for necrotizing enterocolitis, renal dysfunction, hypoglycemia, and increased pulmonary vascular resistance with resultant hypoxia and cyanosis. Although initially thought to cause neurologic dysfunction, the decrease in cerebral blood flow seen in newborns with polycythemia is a physiologic response and does not appear to cause cerebral ischemia.17
Race
In the United States, higher rates of polycythemia vera are observed in the Ashkenazi Jewish population, and lower rates are seen in blacks.
Sex
The male-to-female ratio is 1.2-2.2:1 in adults and 1:1 in children.
Age
The median age for polycythemia vera is 60 years. Only 0.1% of polycythemia cases occur in people younger than 20 years.
Clinical
History
The clinical features associated with polycythemia are a direct result of the increase in red cell mass, which causes an expansion of blood volume. Signs of hyperviscosity and increased metabolism accompany polycythemia. A thorough history must be obtained for a history of cardiac, pulmonary (including sleep apnea), hepatic or renal disease in the patient and a complete family history for evidence of familial polycythemia.
Symptoms include the following:
* Headache
* Weight loss
* Weakness or malaise
* Dizziness
* Pruritus
* Bruising
* Ruddy or red appearance of the skin
* Diaphoresis/dyspnea
* Visual disturbance
* Paresthesias
* Arthropathies
* GI – Fullness, thirst, abdominal discomfort, constipation
Physical
A thorough physical must be completed and include specific evaluation for signs and symptoms of underlying disease that may cause secondary polycythemia; it must include pulse oximetry, careful cardiac and pulmonary evaluation, and evaluation for signs of renal or hepatic disease.
Signs of polycythemia include the following:
* Rubor, especially facial rubor
* Skin plethora
* Hypertension, both systolic and diastolic
* Hepatomegaly
* Splenomegaly
* Conjunctival plethora (engorged vessels in the bulbar conjunctiva)
* Ecchymosis
* Cardiac hypertrophy (rarely observed)
Causes
* Primary polycythemia
o In the past, the pathophysiology was unclear, and primary polycythemias were thought to be due to both inherited and acquired mutations in erythroid progenitors, leading to abnormal red cell proliferation. However, in 2005, an activating mutation found in the tyrosine kinase JAK2 was implicated as the causative factor in polycythemia vera (PV). Five separate groups identified this mutation in approximately 80% (56-97% reported) of patients with polycythemia vera.
o This acquired V617F mutation in JAK2 leads to constitutively activated JAK2. Activated JAK2 induces erythropoietin (Epo) hypersensitivity; although not yet completely delineated, it is thought to act through an activating EPOR.
o Additional JAK2 mutations have been identified in exon 12,9 JAK2H538-K539delinsI ,18 and others.19
o Primary familial and congenital polycythemia (PFCP), which is commonly found to have mutations in the Epo receptor (EPOR) gene. Approximately 14 mutations have been identified.
* Secondary polycythemia
o Congenital causes include high affinity hemoglobin and 2,3-Bisphosphoglycerate (2,3-BPG) deficiency.
o Chuvash polycythemia, a congenital polycythemia first recognized in an endemic Russian population, has mutations in the von Hippel-Lindau (VHL) gene, which is associated with a perturbed oxygen-dependent regulation of Epo synthesis.
o Acquired causes included hypoxemia and Epo-secreting tumors.
o Polycythemia of the newborn usually results from a poor intrauterine environment or hypoxic insult during labor or delivery.
Treatment
Medical Care
* Primary polycythemia: The goals of therapy are to maximize survival while minimizing the complications of therapy as well as of the disease itself.
o Phlebotomy and myelosuppressive chemotherapy are the cornerstones of therapy and have produced a median survival time of 9-14 years after the beginning of treatment. The goal of phlebotomy is to maintain normal red cell mass and blood volume, with a target hematocrit of 45%. The mean survival time of adult patients treated solely with phlebotomy is 13.9 years; however, a high risk of thromboembolic complications is observed.
o In the past, patients have been treated with chlorambucil and other alkylating agents such as pipobroman and busulfan. However, these patients exhibited the highest rates of secondary malignancy including acute leukemia, lymphocytic lymphomas, and skin and GI carcinomas. The rates of malignancy appear lower with busulfan than with the other alkylating agents. Currently, these agents are rarely used.
o Patients treated with phosphorus-32 (32 P) tolerate treatment well and have prolonged periods of remission. However, these patients also exhibit increased rates of acute leukemias (10-15%). The mean survival time with32 P treatment is 10.9 years.
o Studies suggest that the use of interferon alfa decreases the need for phlebotomy and decreases the risk of thrombotic events. Its use is limited by side effects, cost, and route of administration.
o Recent studies using hydroxyurea as a myelosuppressive agent also show promise, reducing the need for phlebotomy. However, similarly to those treated with chlorambucil, these patients also experience higher rates of malignancy. Early clinical studies using imatinib are currently underway and are thus far inconclusive.
o Current recommendations for treatment of young patients rely primarily on phlebotomy because the thrombosis is far less likely to occur in children and the long-term risks of leukemia over a longer life span are increased.
o In the past, the use of anticoagulants, including antiplatelet drugs such as aspirin and dipyridamole (Persantine) had demonstrated increased risk of bleeding without an associated decrease in thrombotic events; therefore, anticoagulants have not previously been recommended. However, a large European study, results of which were published in the New England Journal of Medicine by Landolfi et al (2004),20 showed a decrease in thrombotic events in those patients receiving low-dose aspirin therapy and recommended aspirin therapy for those patients for whom no contraindications existed. This issue continues to remain under debate in the field of polycythemia treatment.
* Secondary polycythemia: Phlebotomy is used for symptomatic hyperviscosity. The goal is to treat the underlying cause of polycythemia.
Surgical Care
Surgery is not typically indicated. Occasionally, splenectomy is performed late in the course of the disease if massive splenomegaly causes adverse effects such as early satiety, anemia, or thrombocytopenia from sequestration.
Please note that these patients have a high risk of complications during surgical procedures.
Consultations
Consult a neurologist and neurosurgeon if evidence of a stroke is present.
Diet
Diet is unrestricted.
Activity
Contact sports and other activities should be limited for individuals in hypercoagulable and hypocoagulable states.
Medication
Current recommendations for treatment of young patients rely primarily on phlebotomy.
Antineoplastic agents
The following medications are not approved for pediatric polycythemia but are extrapolated from other pediatric treatment regimens, including leukemia and myelodysplastic syndrome.
Interferon alfa 2a and 2b (Roferon-A [alfa-2a], Intron A [alfa-2b])
A recombinant purified protein used IV for CML, hairy cell leukemia, and Kaposi sarcoma. Inhibits cellular growth and alters cell differentiation.
Dosing
Adult
CML: 9 million U/d IM/SC; initiate with 3 million U/d, increase by 3 million U every third day; not to exceed 9 million U/d
Pediatric: 2.5-5 million U/d IM/SC
Interactions:Theophylline may increase toxicity; cimetidine may increase antitumor effects; zidovudine and vinblastine may increase toxicity.
Contraindictions : Documented hypersensitivity.
Precautions:
Pregnancy
C – Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus.
Precautions
Caution in brain metastases, severe hepatic or renal insufficiencies, seizure disorders, multiple sclerosis, or compromised CNS; use has been associated with depression, suicidal ideation and suicide attempts, and GI hemorrhage
Chlorambucil (Leukeran)
Antineoplastic alkylating agent of nitrogen mustard type used for CLL, giant follicular lymphoma, Hodgkin lymphoma, and lymphosarcoma.
Dosing:
Adult
0.1-0.2 mg/kg/d PO; adjust dose according to blood count
Pediatric
Not established; limited data available
Intractions:Live virus vaccines (eg, MMR) may result in severe or fatal infection when used in immunosuppressed patients
Contraindictions :Documented hypersensitivity; previous resistance to medication
Precautions:
Pregnancy
D – Fetal risk shown in humans; use only if benefits outweigh risk to fetus
Precautions
Caution in history of seizure disorders or current bone marrow suppression
Busulfan (Myleran)
Potent cytotoxic drug that, at recommended dosage, causes profound myelosuppression. As alkylating agent, mechanism of action of active metabolites may involve cross-linking of DNA, which may interfere with growth of normal and neoplastic cells.
Dosing:
Adult
4-8 mg/d PO; may administer up to 12 mg/d; maintenance dosing range is 1-4 mg/d to 2 mg/wk; discontinue regimen when WBC reaches 10,000-20,000 cells/mL; resume therapy when WBC reaches 50,000/mL
Pediatric
0.06-0.12 mg/kg/d or 1.8-4.6 mg/m2/d PO; titrate dose to maintain WBC >40,000/mL; reduce dose by 50% if WBC is 30,000-40,000/mL; discontinue if WBC <20,000/mL
Pipobroman (Vercyte, Vercite)
The mechanism of action is not fully understood; however, the drug is considered to be an alkylating agent. Pipobroman has been used with some success for treatment of polycythemia vera and chronic granulocytic leukemia. The product was discontinued by the manufacturer in the United States in 1996 but is available in Europe.
Dosing
Adult
1 mg/kg/d PO initially for at least 30 d; if refractory, may increase to 1.5-3 mg/kg/d
Maintenance: 0.1-0.2 mg/kg/d PO; typically initiated when hematocrit has decrease by 50-55%
Pediatric
<15 years: Not established
>15 years: Administer as in adults
Follow-up
Inpatient & Outpatient Medications
* Allopurinol for hyperuricemia or gout
* Iron supplementation to manage the increased red cell production that may produce a functional iron deficiency that can cause red cell rigidity and increase the risk of stroke
* Folate
* Cimetidine for pruritus and upper GI distress
Complications
* Vascular occlusive events – Splenic infarcts, thrombosis (cerebral, portal vein, pulmonary embolus)
* Hemorrhage
* Marrow fibrosis resulting in pancytopenia
* Malignancy – Acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), lymphoma
* Hyperuricemia – Renal stones, nephropathy, gout
* Budd-Chiari syndrome
Prognosis
* The median survival time for patients with polycythemia vera (PV) is 18 months for untreated patients and 9-14 years for treated patients.
Patient Education
* Inform patients that they are prone to surgical complications and are at high risk in trauma situations secondary to coagulopathies.
Miscellaneous
Medicolegal Pitfalls<%2
![Reblog this post [with Zemanta]](https://i0.wp.com/img.zemanta.com/reblog_e.png?w=580)





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}